데이터(Clinical + Omics) 전처리 및 모델링 파이프라인
데이터(Clinical + Omics) 전처리 및 모델링 파이프라인
(Clinical + Omics) 데이터 전처리 및 모델링 파이프라인 보고서
1. 서론
본 보고서는 암 샘플과 정상 샘플을 구별하기 위한 다중 오믹스 데이터 및 임상 데이터를 활용한 머신러닝 모델 개발 과정을 요약합니다. 초기 모델링에서 과적합 문제가 발생하였고, 이를 해결하기 위해 데이터 전처리 방식과 교차 검증 전략을 수정하였습니다. 최종적으로 현실적인 성능 지표를 얻었으며, 그 과정을 단계별로 기술합니다.
2. 데이터 준비 및 전처리
2.1 사용 데이터
- 임상 데이터: 환자 기본 정보, 병기, 수용체 상태 등
오믹스 데이터: 정상(Normal) 및 암(Cancer) 조직에서 추출된 다음 5종류
- 유전자 발현 (Gene Expression - GeneExp)
- 마이크로RNA 발현 (miRNA)
- 복제수 변이 (Copy Number Variation - CNV)
- DNA 메틸레이션 (Methylation - Meth)
- 체세포 돌연변이 (Somatic Mutation - Mut)
2.2 초기 전처리 (오믹스 데이터 공통)
- 환자 ID 통일:
bcr_patient_barcode를 기준으로 대문자 통일 데이터 형태 변환:
- 변이 데이터 → 이진 행렬 (유전자 x 환자)
- 메틸레이션 데이터 → 중앙값으로 결측치 대체
- 발현형 데이터 (GeneExp, miRNA, CNV) → wide format으로 피벗, 결측치는 0으로 대체
후처리 (변이 제외):
- log₂(x+1) 변환 (필요 시)
- 분산 필터링 (하위 5%)
- StandardScaler로 스케일링
- 오믹스 타입 접두사 추가 (
GeneExp_TP53등)
2.3 PCA 적용 방식 (수정)
PCA 과적합 방지 조치:
- 정상/암 데이터를 결합하여 PCA 모델 학습
- 동일한 주성분 공간으로 개별 데이터 변환
메틸레이션 데이터:
- 상위 분산 CpG 사이트 10,000개 선택 후 PCA
2.4 임상 데이터 전처리
- 필요 컬럼만 선택
결측치 처리:
- 수치형 → 중앙값 대치 + StandardScaler
- 범주형 → 최빈값 대치 + OneHotEncoder
- 최종 임상 특징은 환자 ID를 인덱스로 설정
2.5 최종 데이터셋 구축
- 오믹스 특징 취합 (정상/암 샘플 기준)
- 임상 데이터 병합
샘플 리스트 생성:
{'sample_id': '환자ID_NORMAL/CANCER', 'target': 0/1, ...}- Pandas DataFrame 변환 및
sample_id를 인덱스로 설정 - 최종 결측치 처리: SimpleImputer(중앙값)
저장:
final_sample_based_features_clin_omics_revised.csvfinal_sample_based_target_clin_omics_revised.csv
3. 모델링 및 평가
3.1 데이터 로드 및 준비
- 저장된 X, y 파일 로드
- Group 정보 생성 (환자 단위 분할용)
- 타겟 변수 인코딩, 특징 이름 정리 (XGBoost, LightGBM 호환용)
3.2 교차 검증 및 모델 학습
- GroupKFold (5-폴드) 사용
- SMOTE 적용: 소수 클래스에 대해 동적
k_neighbors 사용 모델:
- Logistic Regression (L1)
- Random Forest (max_depth=10 등 과적합 방지)
- XGBoost, LightGBM
- SVM (Linear, RBF)
- Neural Network (MLP)
- 각 모델 학습 및 예측 수행
3.3 모델 평가
- Accuracy, Precision, Recall, F1, ROC AUC
classification_report, 혼동 행렬 출력
3.4 특징 중요도 분석
- 모델별 고유 중요도 (feature_importances_, coef_ 등)
SHAP 분석:
- TreeExplainer, LinearExplainer, KernelExplainer 사용
- 폴드 평균값으로 중요도 시각화
3.5 PCA 컴포넌트 해석
- SHAP 상위 PCA 특징 해석
- 주성분 로딩값 시각화 (각 오믹스 원본 특징 기반)
4. 결과 및 논의
4.1 초기 과적합 문제
- 모델 성능이 ROC AUC 등에서 1.0 근접 → 과적합 발생
원인:
- StratifiedKFold 사용 → 데이터 유출
_normal/_cancer접미사 → 클래스 정보 유출
4.2 해결 노력
- GroupKFold로 교차 검증 전략 변경
전처리 로직 수정:
- 접미사 제거
- PCA 방식 수정
- 임상 데이터 일시 제거 후 재포함하여 문제 확인
4.3 최종 성능 요약
| 모델 | Accuracy | Precision | Recall | F1 | ROC AUC |
|---|---|---|---|---|---|
| Logistic Regression (L1) | 0.319 | 0.411 | 0.319 | 0.289 | 0.195 |
| Random Forest | 0.877 | 0.896 | 0.877 | 0.868 | 0.835 |
| XGBoost | 0.868 | 0.878 | 0.868 | 0.860 | 0.822 |
| LightGBM | 0.858 | 0.862 | 0.858 | 0.850 | 0.848 |
| SVM (Linear) | 0.323 | 0.415 | 0.323 | 0.300 | 0.800 |
| SVM (RBF) | 0.735 | 0.741 | 0.735 | 0.737 | 0.757 |
| Neural Network (MLP) | 0.358 | 0.599 | 0.358 | 0.262 | 0.304 |
- 과적합 문제 해결 → 현실적인 성능 도출
- 트리 기반 모델(RF, XGB, LGBM)이 상대적으로 우수
5. 결론 및 향후 계획
- 데이터 유출 방지를 위한 전처리 및 교차 검증 전략 수정 → 과적합 해결
향후 개선 방향:
- 하이퍼파라미터 최적화 (GridSearchCV 등)
- 중요 특징 심층 분석 및 해석 (SHAP, 생물학적 해석)
- 도메인 지식을 활용한 특징 선택/공학
- 외부 데이터셋 검증으로 일반화 성능 확인
6. 결과 사진
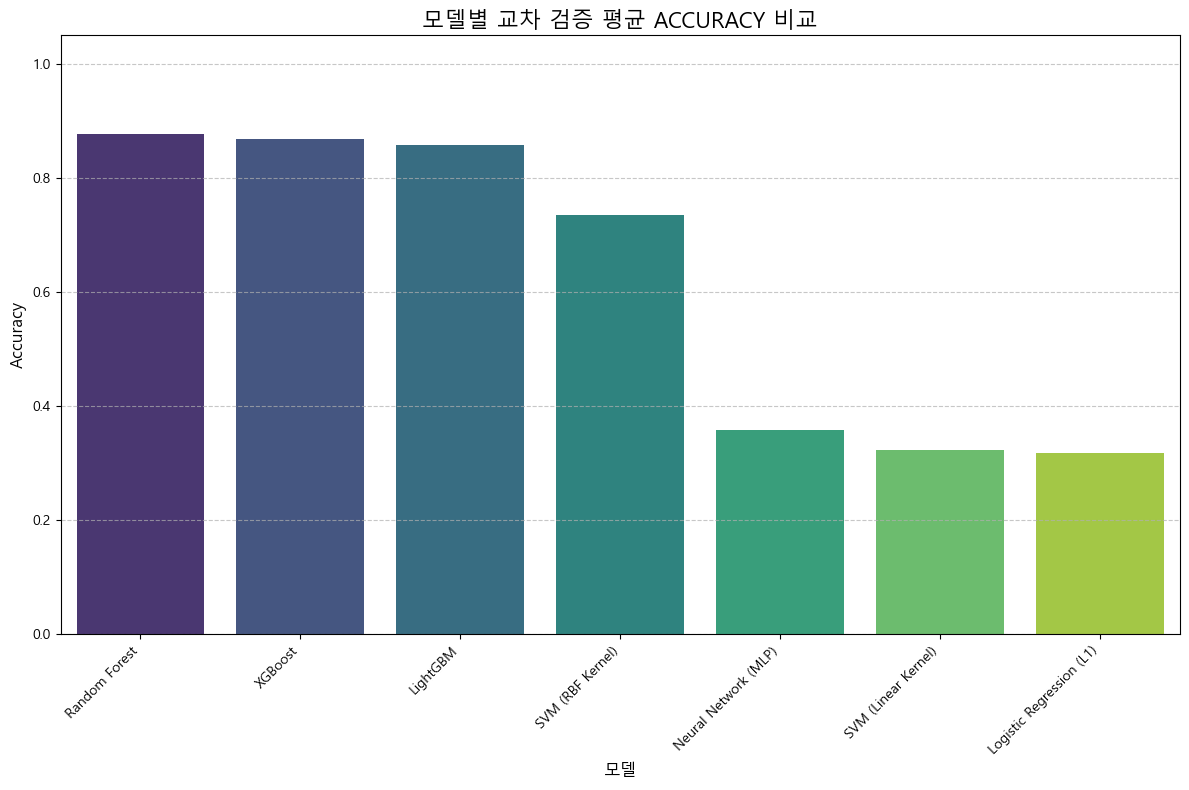
모델 성능 비교
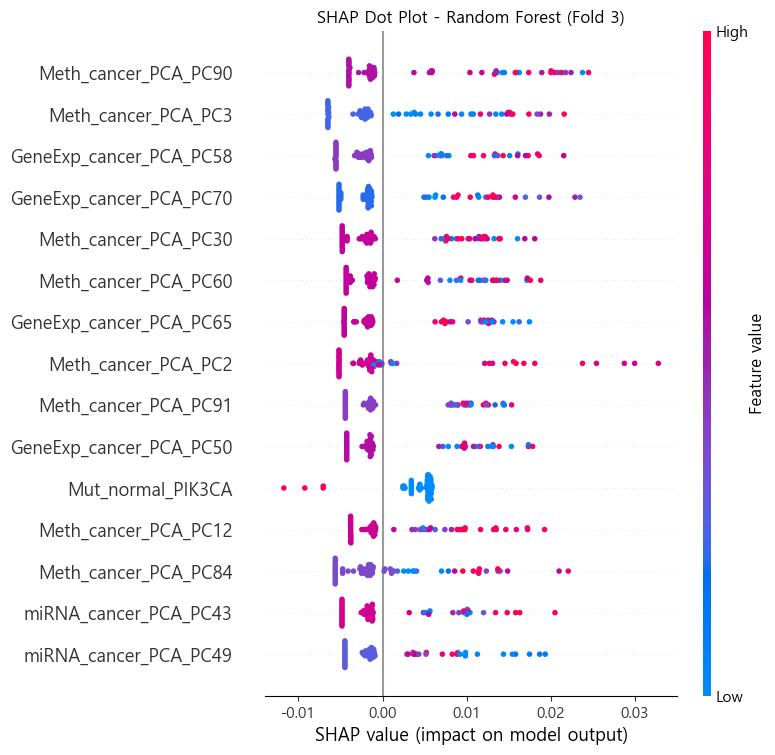
중요 바이오마커 예시(가장 높은 성능의 Random Forest_fold3)
This post is licensed under CC BY 4.0 by the author.